Charge neutrality#
where \([D_q]\) is the concentration of defect \(D\) in charge state \(q\), \(n_e\) is the concentration of electrons and \(n_h\) is the concentration of holes. At a given temperature \(T\), we can solve this equation and determine the Fermi level position and the equilibrium concentrations.
In defermi, the equilibrium Fermi level can be computed with the solve_fermi_level function. Let’s see how it works with our usual example.
[1]:
import pandas as pd
from defermi import DefectsAnalysis
bulk_volume = 800 # cubic Amstrong
data = [
{'name': 'Vac_O','charge': 2,'multiplicity': 1,'energy_diff': 7,'bulk_volume': bulk_volume},
{'name': 'Vac_O','charge':0,'multiplicity':1,'energy_diff': 10.8, 'bulk_volume': bulk_volume},
{'name': 'Vac_Sr','charge': -2,'multiplicity': 1,'energy_diff': 8,'bulk_volume': bulk_volume},
{'name': 'Vac_Sr','charge': 0,'multiplicity': 1,'energy_diff': 7.8,'bulk_volume': bulk_volume},
]
df = pd.DataFrame(data)
da = DefectsAnalysis.from_dataframe(df,band_gap=2.0,vbm=0.0)
chempots = {'O':-5,'Sr':-2}
da.plot_formation_energies(chempots,figsize=(4,4));
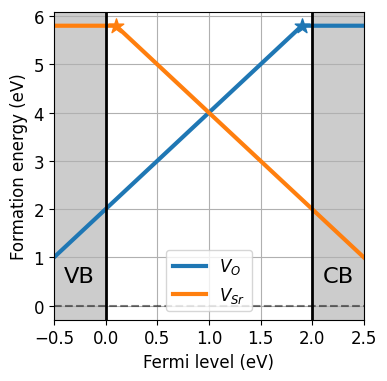
[12]:
# Find equilibrium Fermi level solving charge neutrality
da.solve_fermi_level(
chemical_potentials=chempots,
bulk_dos={'m_eff_e':0.5,'m_eff_h':0.4},
temperature=1000)
[12]:
0.9855782323977027
The bulk_dos argument is needed for the calculation of the electronic carriers (electrons and holes), which is the focus of the next section.
Electronic carriers#
The concentrations of intrinsic carriers are derived by integrating the number of unoccupied states up to the VBM (\(\mathrm{E_{VBM}}\)) for holes:
and the number of occupied states from the CBM (\(\mathrm{E_{CBM}}\)) for electrons:
We can also express it in terms of effective masses:
DOS#
In defermi, both the numerical integration using the density of states (DOS, \(D(\epsilon)\)) and the effective mass approximation are available. If the DOS is available from a VASP calculation, the easiest way is to pass a pymatgen Dos (or FermiDos or CompleteDos) object. If not available, it also can be pulled from the Materials Project Database.
[9]:
from defermi.tools.materials_project import MPDatabase
bulk_dos = MPDatabase().get_dos_from_stable_composition('SrO')
bulk_dos
[9]:
<pymatgen.electronic_structure.dos.CompleteDos at 0x7834c2b94050>
If a DOS from any other DFT code is available, you can pass it as a dict, with the following keys and values:
"energies":listornp.arraywith energy values"densities":listornp.arraywith total density values"structure": pymatgenStructureof the material, needed for DOS volume and charge normalization
In both cases, the integration is carried out using pymatgen’s FermiDos class.
Effective masses#
If the DOS is not available, you can provide the effective masses of electrons and holes as a dictionary: {"m_eff_e":value, "m_eff_h":value}, as in the previous example. The number of carriers is computed as explained above.
Chemical potentials#
The chemical potentials are defined in the same way as for Formation energies (with the possibility to pull values from the MP database).
Self-consistent solution#
The charge neutrality equation is solved using scipy’s bisect method, with \(\epsilon_F\) range \([-1,E_g + 1]\), with \(\epsilon_F\) set to zero at \(E_{\mathrm{VBM}}\).