Custom energies and concentrations#
defermi offers the possibility to customize the core functions DefectEntry.formation_energy and DefectEntry.defect_concentration. This is especially useful whenever we want to simulate conditions that deviate from the standard ideal solution model, like temperature-dependent formation energies, or anisotropic systems like interfaces and grain boundaries. In this example, we implement temperature-dependent formation energies for \(V_O\).
Temperature-dependent formation energies#
We start from our usual example with Sr and O vacancies.
[39]:
import pandas as pd
from defermi import DefectsAnalysis
import matplotlib.pyplot as plt
import seaborn as sns
sns.set_theme(context='talk',style='ticks')
bulk_volume = 800 # cubic Amstrong
data = [
{'name': 'Vac_O','charge': 2,'multiplicity': 1,'energy_diff': 7,'bulk_volume': bulk_volume},
{'name': 'Vac_O','charge':0,'multiplicity':1,'energy_diff': 10.8, 'bulk_volume': bulk_volume},
{'name': 'Vac_Sr','charge': -2,'multiplicity': 1,'energy_diff': 8,'bulk_volume': bulk_volume},
{'name': 'Vac_Sr','charge': 0,'multiplicity': 1,'energy_diff': 7.8,'bulk_volume': bulk_volume},
]
df = pd.DataFrame(data)
da = DefectsAnalysis.from_dataframe(df,band_gap=2.0,vbm=0.0)
bulk_dos={'m_eff_e':0.5,'m_eff_h':0.4}
chempots = {'Sr':-2,'O':-5}
da.plot_formation_energies(chempots,figsize=(4,4));
plt.show()
precursors = {'SrO':-10}
da.plot_brouwer_diagram(bulk_dos=bulk_dos,temperature=1000,precursors=precursors,
oxygen_ref=-5,pressure_range=(1e-35,1e25),
figsize=(4,4),fontsize=14);
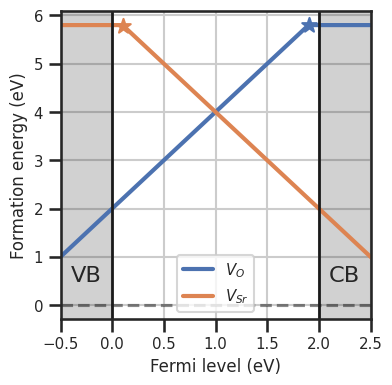
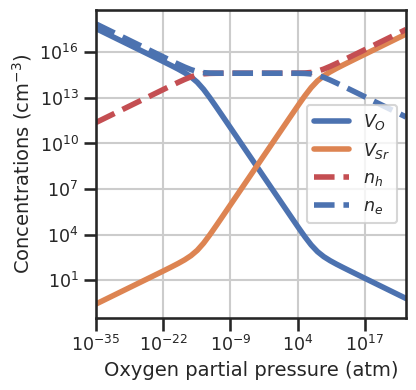
Let’s assume we were able to calculate the dependency of the formation energies on temperature. We can now define a function to compute the formation energy for this specific case. The function inputs need to be the same as the inputs of the default formation_energy function, plus the custom **kwargs:
entry:DefectEntryobjectvbmchemical_potentialsfermi_leveltemperature**kwargs
In this example, we compute the T-dependent formation energy as:
with:
For the formation energy at \(0 K\) use use the _default_formation_energy_function, but it could be programmed explicitely if needed.
[26]:
# T-dependent formation energy of VO (made-up for example)
def formation_energy_Vac_O(
entry,
vbm=None,
chemical_potentials=None,
fermi_level=0,
temperature=0,
**kwargs):
formation_energy = entry._default_formation_energy(vbm=vbm,chemical_potentials=chemical_potentials,fermi_level=fermi_level)
return formation_energy - 1/1500 *temperature
# T-dependent formation energy of VSr (made-up for example)
def formation_energy_Vac_Sr(
entry,
vbm=None,
chemical_potentials=None,
fermi_level=0,
temperature=0,
**kwargs):
formation_energy = entry._default_formation_energy(vbm=vbm,chemical_potentials=chemical_potentials,fermi_level=fermi_level)
return formation_energy + 1/1100 *temperature
import numpy as np
import matplotlib.pyplot as plt
fig,ax = plt.subplots(figsize=(4,4))
temperatures = np.linspace(0,1000,100)
entry = da.select_entries(name='Vac_O',charge=2)[0]
Y = [formation_energy_Vac_O(entry=entry,vbm=0,chemical_potentials=chempots,fermi_level=1,temperature=T) for T in temperatures]
ax.plot(temperatures,Y,lw=3,label=entry.symbol_charge)
entry = da.select_entries(name='Vac_Sr',charge=-2)[0]
Y = [formation_energy_Vac_Sr(entry=entry,vbm=0,chemical_potentials=chempots,fermi_level=1,temperature=T) for T in temperatures]
ax.plot(temperatures,Y,lw=3,label=entry.symbol_charge)
ax.set_xlabel('Temperature (K)')
ax.set_ylabel('$\Delta E_f$ (eV)')
ax.set_xlim(0,1000);
plt.legend();
plt.grid();
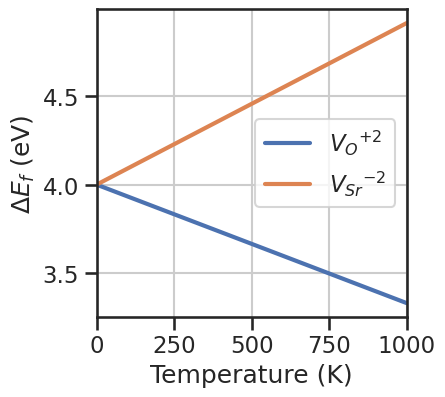
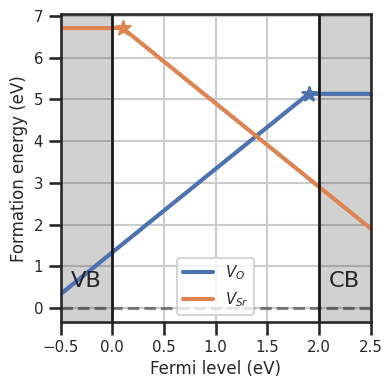
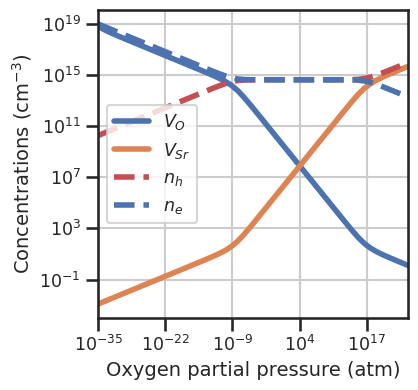
Once the function is defined, we can set the DefectEntry.formation_energy function for the desired defects to our custom function, using the set_formation_energy_functions method. Alternatively we can use directly DefectEntry.set_formation_energy_function for the individual entry. We pass the kwargs to select_entries to choose which entries to set the custom functions. If we plot the formation energies at 1000 K we see that formation energies for \(V_O\) decrease wrt to
the 0 K case, while the \(V_{Sr}\) increase. Also in the Brouwer diagram the balance shifts in favor of \(V_O\).
[41]:
# set formation energies functions for Vac_O
da.set_formation_energy_functions(function=formation_energy_Vac_O,name='Vac_O')
# set formation energies functions for Vac_O
da.set_formation_energy_functions(function=formation_energy_Vac_Sr,name='Vac_Sr')
da.plot_formation_energies(chempots,temperature=1000,figsize=(4,4));
plt.show()
da.plot_brouwer_diagram(bulk_dos=bulk_dos,temperature=1000,precursors=precursors,
oxygen_ref=-5,pressure_range=(1e-35,1e25),
figsize=(4,4),fontsize=14);
Volume-dependent concentrations#
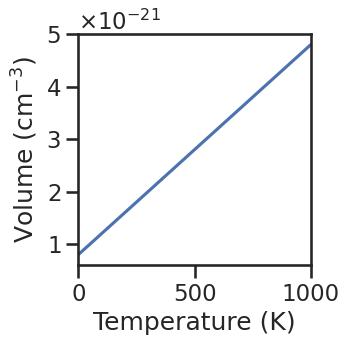
In the next example, we include the temperature-dependence of the cell volume in the defect site multiplicity, which in turn affects the defect concentrations.
[77]:
# T-dependence in volume in defect concentrations (made-up numbers for example)
def defect_concentrations_volume(
entry,
vbm=0,
chemical_potentials=None,
temperature=0,
fermi_level=0,
per_unit_volume=True,
eform_kwargs={},
**kwargs):
# include formation energy kwargs
conc = entry._default_defect_concentration(
vbm=vbm,
chemical_potentials=chemical_potentials,
temperature=temperature,
fermi_level=fermi_level,
per_unit_volume=per_unit_volume,
eform_kwargs=eform_kwargs)
if per_unit_volume:
n_corr = (1 + 1/200*temperature)
return conc * n_corr
else:
return conc
def V(T):
return bulk_volume*(1+1/200*T)*1e-24
plt.figure(figsize=(3,3))
plt.plot(temperatures,[V(T) for T in temperatures])
plt.xlim(0,1000)
plt.xlabel('Temperature (K)')
plt.ylabel('Volume (cm$^{-3}$)')
plt.ticklabel_format(axis='y',style='sci',scilimits=[0,1],useMathText=True)
[ ]:
# set for all entries
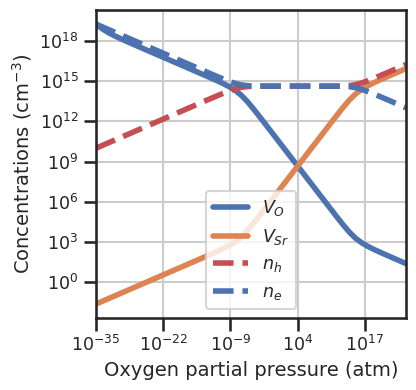
da.set_defect_concentration_functions(function=defect_concentrations_volume)
da.plot_brouwer_diagram(bulk_dos=bulk_dos,temperature=1000,precursors=precursors,
oxygen_ref=-5,pressure_range=(1e-35,1e25),
figsize=(4,4),fontsize=14);
[ ]: